In Vivo Adenine Base Editing Rescues Adrenoleukodystrophy in a Humanized Mouse Model
Jessica Schneller, PhD - August 08, 2024
In this study, Gopalappa et al. sought to precisely correct the pathogenic p.G512S mutation, a frequently observed mutation in patients, using either an adenine base editor (ABE) or a prime editor (PE).
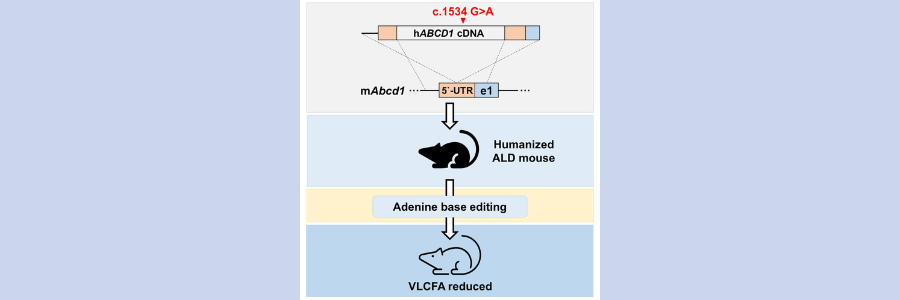
X-linked adrenoleukodystrophy (ALD) is an inherited neurometabolic disorder caused by mutations in ABCD1, a peroxisomal ABC transporter. In ALD patients, very-long-chain fatty acids (VLCFAs) fail to enter the peroxisome and undergo subsequent b-oxidation, resulting in accumulation. Accumulated VLCFAs can cause pathology, such as cerebral inflammation or demyelination in the central nervous system (CNS). While current therapeutic approaches have focused on inhibiting VLCFA accumulation, these treatments do not provide permanent relief. Conventional gene therapy does not fundamentally correct the pathogenic mutation, and depending on the viral vector used, the viral genome can randomly integrate causing tumorigenesis. In this study, Gopalappa et al. sought to precisely correct the pathogenic p.G512S mutation, a frequently observed mutation in patients, using either an adenine base editor (ABE) or a prime editor (PE). To do this, they generated a novel humanized mouse model of ALD (hALD) in which the hABCD1 cDNA harbors the p.G512S mutation, and demonstrated that systemic delivery of NG-ABE8e(V106W) via dual AAV-PHP.B vectors into the hALD mouse could achieve efficient mutation correction and reduced plasma VLCFA levels in vivo. The results suggest that a dual AAV-PHP.B-split-NG-ABE8e(V106W) system could be a promising therapeutic modality for ALD patients.
The authors’ first objective was to generate a humanized ALD mouse model by inserting a hABCD1 cDNA sequence containing the p.G512S mutation into the 5’-UTR of the Abcd1 murine gene using targeted integration. Cas9 mRNA & sgRNA targeting the 5’-UTR of the mouse Abcd1, & double-stranded donor cDNA containing the mutant hABCD1-cDNA were injected into C57BL/6N mouse zygotes; after which zygotes were transplanted into surrogate mothers to obtain offspring. ABCD1 is X-linked & predominantly affects males; thus, male mice positive for the cDNA knock-in were chosen for further analysis and were referred to as hALD mice. The expression of hABCD1 cDNA by quantitative real-time PCR (qRT-PCR) in the brain of hALD mice showed significantly higher expression of the mutated human ABCD1 mRNA in hALD than in wild-type (WT) male mice, and there was a significant increase of VLCFAs in several tissues.
With the goal of correcting the p.G512S mutation in vivo, the authors next aimed to evaluate the therapeutic genome editing efficiencies of CRISPR-Cas9-based prime editing (NGG-PE2max) and base editing (NG-ABE8e(V106W)) modalities in patient-derived fibroblasts. Top-performing prime editing gRNAs (pegRNAs) were identified and screened using a high-throughput method, in which individual pegRNAs were identified by barcodes to determine the optimal spacer, reverse transcriptase template, and primer binding sites for the pegRNAs. pegRNA library constructs were generated in lentivirus libraries, which were tested in HEK293T cells. The screen designated three top-ranked pegRNAs, but when plasmids expressing the pegRNAs were electroporated into patient fibroblasts harboring the mutation along with a plasmid expressing PE2, only modest (<1%) gene correction was observed. Therefore, the authors considered a base editing strategy, which might be more efficient at correcting the mutation in vivo.
In order to develop an adenine base editing system, three different sgRNAs were tested for the ABE variant NG-ABE8e(V106W). These sgRNAs shared the same spacer sequence but with either one or two additional guanine nucleotides incorporated at the 5’ end of the spacer, to enhance expression from the U6 promoter. Because the NG-ABE8e(V106W) and sgRNA sequences exceeded the cargo capacity of a single adeno-associated viral (AAV) vector, the NG-ABE8e(V106W) was split into two halves and intein-mediated trans-splicing was used to reconstitute the full-length protein. To test the editing efficiency of NG-ABE8e(V106W) with the three different sgRNA sequences, two AAV packaging plasmids containing (AAV-N-intein_NG-ABE8e(V106W) and AAV-C-intein_NG-ABE8e(V106W) with sgRNA) were co-transfected into patient-derived fibroblasts. The mutation correction rate was determined by targeted deep sequencing; among the three sgRNA variants, the sgRNA with the addition of a single guanine nucleotide at the 5’-end was associated with the highest editing efficiency (7.44%). Potential off-target sites for this sgRNA were identified using Cas-OFFinder; of the eight sites, only one showed measurable base editing of (~1%) in a non-coding region. Between the two genome-editing approaches, NG-ABE8e(V106W) showed more efficient editing than PE2max; therefore, NG- ABE8e(V106W) was selected for in vivo genome editing.
To evaluate gene correction with the NG-ABEe (V106W)) using the selected sgRNA in hALD mice, components were injected via systemic delivery of AAV. The chosen AAV serotype was PHP.B, a neurotropic AAV evolved from AAV9, which was selected for its high neurotropism and efficient delivery to the brain. The split version of NG-ABE8e(V106W) and sgRNA (AAV-PHP.B-N-intein_NG-ABE8e(V106W) and AAV-PHP.B-C-intein_NG-ABE8e(V106W) with sgRNA) was delivered in 6-week-old hALD mice via tail vein injection. To evaluate the dose dependence of genome editing and subsequent therapeutic effects, two doses of AAV were administered, a high-dose group (6.4e12vg) and a low-dose group (1e12vg). Six weeks later, treated mice were euthanized, and organs were harvested for genome editing. Targeted deep sequencing analysis was conducted to compare base-editing efficiencies at the genomic DNA and RNA levels in multiple organs. The average efficiencies of genomic DNA and RNA editing in the brains of hALD mice systemically administered high-dose AAV were 1%–5.5% and 2.4%–10%, respectively. For mice administered low-dose AAV, the average efficiencies in the brain were 0.7%–2% for genomic DNA and 0.4%–3.2% for RNA. Levels of plasma VLCFAs, mainly C26:0/C22:0 and C26:0, were significantly reduced; both high- and low-dose AAV led to a significant reduction in the C26:0/C22:0 ratio in the plasma, with 49% and 36% reductions from the maximal level in the high- and low-dose AAV groups, respectively. However, there was no noticeable reduction in levels of the C26:0 VLCFA in the plasma.
In conclusion, the authors generated a humanized mouse model of ALD which demonstrated key characteristics of the ALD phenotype, in particular elevated levels of VLCFA. They were able to optimize split-NG-ABE8e(V106W)-sgRNA variants for in vivo therapeutic genome editing through in vitro experiments using ALD patient-derived fibroblasts. Finally, AAV-PHP.B-mediated systemic delivery of the dual split-NG-ABE8e(V106W) in hALD mice showed robust gene editing of the p.G512S mutation in different parts of the brain and in multiple organs, with a reduction of plasma VLCFA. These findings facilitate the clinical translation of the ABE system for permanent correction of this and other ALD mutations, as a treatment for this disorder.
Dr. Schneller is scientist II, gene editing, at ReCode Therapeutics and Associate Editor of The Vector.
Related Articles