The Vector
Volume 7 Issue 11: November 2018
Editorial Team
Phillip Doerfler, PhD - Editor, The Vector
Melvin Rincon, MD, PhD - Associate Editor, The Vector
Edith Pfister, PhD - Junior Editor, The Vector
Inside This Issue
President's Message
Breaking Through
Society News
Public Policy
Industry News
Board of Directors' Message
ASGCT Giving Tuesday and It's (Probably) Time to Renew Your Membership

Since my introduction to the ASGCT in 2010, I have been blown away by the momentum of development and progress in the field. I was privileged to sit on the inaugural New Investigator Committee in 2011, and to see firsthand just how important the role of the Society is in mobilizing key stakeholders—not just researchers, but also patient advocacy groups, government bodies, and industry. The Society’s efforts to engage and educate the public, as well as policy makers and regulators, have been key to strengthening the future of the field. I feel honored to now sit on the Board of Directors at such a marvelous time and am grateful to those mentors and innovators who carved the path before me, continue to engage in new directives and muscle through the obstacles ahead.
Don’t miss out on your chance to be part of our collective progress. I encourage all of our members to ensure their dues are up-to-date and to renew their commitment to ASGCT if their membership is set to expire at the end of the year.
Finally, November 27 is Giving Tuesday—millions of people worldwide use the day to refocus their holiday spirit and determine their strategy for end-of-year charitable contributions. Because of our membership dues and charitable donations, ASGCT has made landmark progress in our advocacy and regulatory efforts in 2018, and, the Society will be launching a brand-new patient education program to be used by patients, clinicians, and patient advocacy groups in early 2019.
Consider Including ASGCT in
Your End-of-Year Giving
Every act of generosity counts and it means even more when our members work together. Join us in supporting both the research that drives our innovations and the patients we design them for by including ASGCT in your Giving Tuesday plans, and share your support with friends by tagging @ASGCTherapy and using #GivingTuesday in your posts.
Between our continued growth in membership and the contributions from our community, there’s no limit to what ASGCT can accomplish.
Best,
Jennifer Adair, Ph.D.
ASGCT Board of Directors

Breaking Through
Therapeutic Genome Editing for Myotonic Dystrophy Type 1 Using CRISPR/Cas9
Yanlin Wang, Lei Hao, Hongcai Wang, Katherine Santostefano, Arjun Thapa, John Cleary, Hui Li, Xiuming Guo, Naohiro Terada, Tetsuo Ashizawa, Guangbin Xia
Summary by Guangbin Xia, M.D., Ph.D.
DOI: https://doi.org/10.1016/j.ymthe.2018.09.003
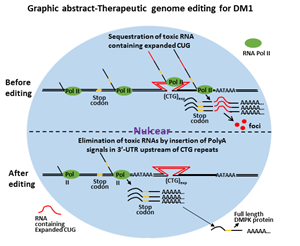 Myotonic dystrophy type 1 (DM1) is a dominant monogenic neurodegenerative disorder, and it is the most common muscular dystrophy in adults1. The prevalence of DM1 is 8-10/100,0002 and has been described in people from all over the world. Its congenital form, congenital myotonic dystrophy, is reported to have an incidence of 2.1 per 100,000 live birth3. It is a fatal disease. The main causes of morbidity and mortality in adult are from progressive muscle wasting, respiratory failure, sudden cardiogenic death and central nervous system involvement1, 4-6. CDM has 30-40% neonatal mortality7. Surviving infants experience developmental delay, cognitive impairment, psychiatric issues, gastrointestinal dysfunction, and cardiac arrhythmias8 and eventually succumb to progressive muscle wasting before 40s. However, currently, there is no effective treatment.
Myotonic dystrophy type 1 (DM1) is a dominant monogenic neurodegenerative disorder, and it is the most common muscular dystrophy in adults1. The prevalence of DM1 is 8-10/100,0002 and has been described in people from all over the world. Its congenital form, congenital myotonic dystrophy, is reported to have an incidence of 2.1 per 100,000 live birth3. It is a fatal disease. The main causes of morbidity and mortality in adult are from progressive muscle wasting, respiratory failure, sudden cardiogenic death and central nervous system involvement1, 4-6. CDM has 30-40% neonatal mortality7. Surviving infants experience developmental delay, cognitive impairment, psychiatric issues, gastrointestinal dysfunction, and cardiac arrhythmias8 and eventually succumb to progressive muscle wasting before 40s. However, currently, there is no effective treatment.
DM1 is caused by CTG nucleotide repeat expansion within the 3 prime untranslated region (3’-UTR) of the Dystrophia Myotonica Protein Kinase (DMPK) gene9. The expanded CTG repeats encode toxic CUG RNA that causes disease largely through RNA gain-of-function2, 10-13. With the advancement of therapeutic genome-editing technologies, it is becoming more realistic to correct or edit the mutant genome so that the disease can be fundamentally cured. In order to eliminate the toxic RNA CUG repeats, we first tried to delete the entire CTG repeats using CRISPR/Cas9. We found paired SpCas9 or SaCas9 guide RNA flanking the CTG repeats induced deletion of expanded CTG repeats and elimination of toxic RNA CUG repeats. However, this approach also incurred frequent inversion of the flanked sequences. The inversion caused the transcription of DMPK gene with an expanded RNA CAG repeats, which were still trapped in the nuclei. The deleterious effects of the inversion remain undetermined. Other genetic events related to targeted deletion are unpredictable. We caution the use of this approach for in vivo therapeutic genome editing.
We then attempted the insertion of polyadenylation signals in the 3’-UTR upstream of the CTG repeats to eliminate toxic RNA CUG repeats (graphic abstract). This was achieved by precise integration through homology-directed repair facilitated by a pair of gRNA/SpCas9 nickase. The strategy was tested on DM1 induced pluripotent stem cells (iPSCs), a cellular model of DM1. We found the DMPK transcription was prematurely terminated following the engineered polyadenylation signals, which generated DMPK pre-mRNA with all exons but lack the expanded CUG repeats that define mutant DMPK. We have found the post-transcription procession of these mRNA precursors was very similar to the wild type DMPK. This is particularly suitable for in vivo therapeutic development because the event can be precisely predicted and the DMPK function would not be affected even if both the mutant and the normal allele were targeted. The manipulation of the genome did not affect the pluripotency of the iPSCs. We have found the elimination of toxic RNA CUG repeat foci and reversal of alternative splicing in differentiated neural stem cells, cardiomyocytes, and skeletal muscle myofibers. This approach not only eliminated the toxic CUG repeats but also released newly engineered DMPK transcripts into the cytoplasm, which could potentially offset abnormalities that are caused by haploinsufficiency. We concluded that targeted insertion of polyadenylation signals into the 3’-UTR, upstream of expanded CTG repeats, is a viable approach for the development of therapeutic genome editing for DM1. The direct clinical application for this type of therapeutic genome editing will be the development of personalized cell-based therapy. The genome-edited patient-specific iPSCs can be differentiated into skeletal muscle progenitor cells, which can be used for cell replacement therapy for advanced DM1. This approach may also be applied to in vivo therapeutic genome editing before the onset of the disease or for congenital myotonic dystrophy.
References
- Harper P. Myotonic Dystrophy, 3rd ed. London: WB Saunders, 2001
- Romeo V. Myotonic Dystrophy Type 1 or Steinert's disease. Adv Exp Med Biol. 2012;724:239-257
- Campbell C, Levin S, Siu VM et al. Congenital myotonic dystrophy: Canadian population-based surveillance study. J Pediatr. 2013;163:120-125 e121-123
- Bassez G, Lazarus A, Desguerre I et al. Severe cardiac arrhythmias in young patients with myotonic dystrophy type 1. Neurology. 2004;63:1939-1941
- Groh WJ, Groh MR, Saha C et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008;358:2688-2697
- Harper P. Major problems in neurology, in Myotonic Dystrophy(ed3).
- Echenne B, Rideau A, Roubertie A et al. Myotonic dystrophy type I in childhood Long-term evolution in patients surviving the neonatal period. Eur J Paediatr Neurol. 2008;12:210-223
- Reardon W, Newcombe R, Fenton I et al. The natural history of congenital myotonic dystrophy: mortality and long term clinical aspects. Arch Dis Child. 1993;68:177-181
- Fu YH, Pizzuti A, Fenwick RG, Jr. et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256-1258
- Ashizawa T, Sarkar PS. Myotonic dystrophy types 1 and 2. Handb Clin Neurol. 2011;101:193-237
- Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259-277
- Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans. 2009;37:1281-1286
- Gomes-Pereira M, Cooper TA, Gourdon G. Myotonic dystrophy mouse models: towards rational therapy development. Trends Mol Med. 2011;17:506-517
Society News

Abstract Submission is Now Open
With the opening of abstract submission for the 22nd Annual Meeting, we’re officially in #ASGCT19 season. Submit an abstract by January 16 to be eligible for free annual meeting registration, travel awards, and Excellence in Research awards.
Submit an Abstract Online
Full Summary of ASGCT's FDA Liaison Meeting
 Six field-leading experts presented as part of the first-ever ASGCT liaison meeting with the FDA on September 13, 2018. The meeting covered Society recommendations to FDA CBER on manufacturing, safety, and testing requirements for gene therapies, as well as an FDA update on the new RMAT designation. This deep-dive longread includes the recommendations from those discussions.
Six field-leading experts presented as part of the first-ever ASGCT liaison meeting with the FDA on September 13, 2018. The meeting covered Society recommendations to FDA CBER on manufacturing, safety, and testing requirements for gene therapies, as well as an FDA update on the new RMAT designation. This deep-dive longread includes the recommendations from those discussions.
Meeting Agenda and Presentations
are Available Online
Public Policy
ASGCT White Paper Published in Molecular Therapy
The latest ASGCT, Addressing the Value of Gene Therapy and Enhancing Patient Access to Transformative Treatment, was published on the Molecular Therapy website and will be featured in The December issue, reflecting the journal's new commitment to cover not only cutting-edge science, but also pressing policy issues. The paper highlights the significance of addressing challenges that could affect patient access to the substantial value of approved gene therapies and identifies support for proposed policy solutions—the creation of new reporting and billing codes and improved Medicare reimbursement levels for these therapies.
Sickle Cell Disease Bill Passes Senate
Last month, the Senate passed S. 2465, the Sickle Cell Disease (SCD) and Other Heritable Blood Disorders Research, Surveillance, Prevention and Treatment Act of 2018. Through sign-on letters, ASGCT has supported this legislation’s proposed authorization of a national surveillance program for SCD and other heritable blood disorders and reauthorization of an SCD treatment demonstration program. A companion bill previously passed the House, but the differing Senate version of the bill will go back to the House for a final vote.
Industry News
Interested in advertising in The Vector?
View the Rate Sheet